Note (GPU cards are the same type as on Karolina supercomputer at IT4Innovations)
The advance of computing power has opened new perspectives of computational materials design. Accurate predictions of the intrinsic physical properties are routinely obtained by performing ab-initio simulations at the atomistic level. These calculations, however, have severe restrictions on the size and temporal evolution of the systems. Finite-temperature properties of nano- and meso-scale systems (104 to 107 atoms) are simulated via classical Molecular Dynamics (MD). The atomic interactions are described by parametrized force-fields, which lack the transferability and desired accuracy over different temperature and pressure conditions. A reactive force-field potential, ReaxFF (https://en.wikipedia.org/wiki/ReaxFF), offers a trade-off between accuracy and system size. ReaxFF employs a bond-order formalism in conjunction with polarizable charge descriptions to describe both reactive and non-reactive interactions between atoms [1]. This allows ReaxFF to accurately model both covalent and electrostatic interactions for a diverse range of materials.
There is an increased trend of using Graphical Processing Units (GPUs) in modern supercomputers. A version of ReaxFF is implemented in one of the widely used MD codes LAMMPS (https://www.lammps.org/). LAMMPS implementation offers the possibility of parallel execution of ReaxFF on GPUs through KOKKOS libraries. We have recently performed a scaling test on the Jewels Booster Module, one of the powerful supercomputers (https://apps.fz-juelich.de/jsc/hps/juwels/configuration.html) in Julich, and with a configuration similar to IT4I supercomputers: 2×AMD EPYC Rome 7402 CPU (2×24 cores) equipped with 4×NVIDIA A100 GPU (4×40 GB). We used LAMMPS stable release, 29Oct20 version, compiled with CUDA aware OpenMPI (GCC 10.3.0, CUDA 11.3, OpenMPI 4.1.1) and including the following packages: : Body, Kspace, Manybody, Phonon, Qeq, Reaxff, and Kokkos. Calculations were performed with 1 MPI process per GPU (4 MPI processes per node). OpenMP threads were not used. The input data were taken from LAMMPS benchmarks (bench_reaxc.tar.gz from https://www.lammps.org/bench.html). We ran 1000 iterations with 0.1fs timestep for each calculation. In Fig.1 we show the strong scaling for different system sizes.
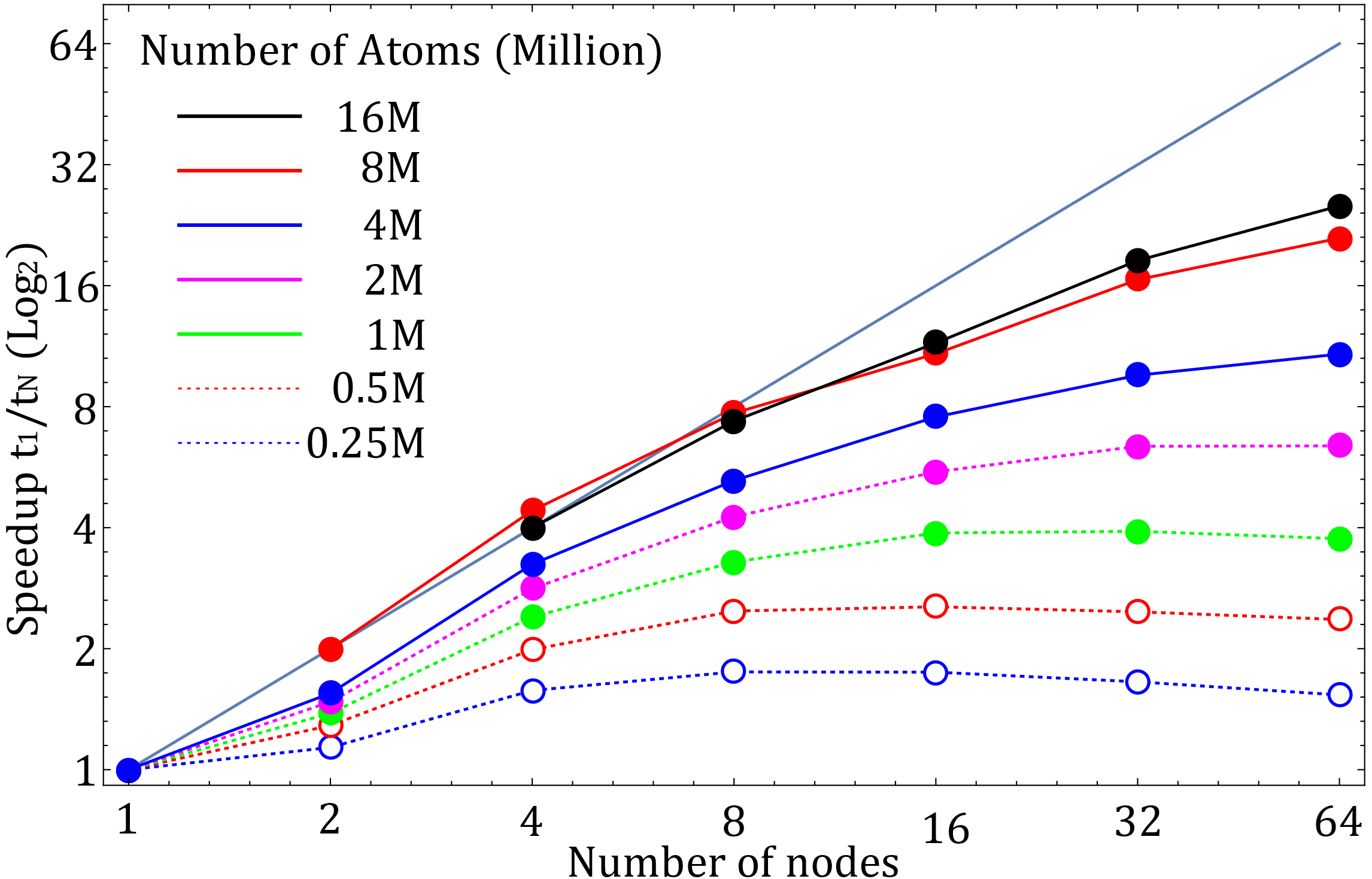
Figure 1. Strong scaling for cases with different number of atoms (both axes show the data in base 2 logarithm). Test case with 8 million atoms did not fit on single node, and the one with16 million atoms did not fit on 2 nodes. In these two cases, t1 was extrapolated by assuming an ideal scaling.
The strong scaling shows that the efficiency of parallel execution on GPUs when using ReaxFF increases with the size of the system. Thus, for the best performance, one should consider running calculations for systems with 100 of millions of atoms.
In Fig.2 we also show the weak scaling. From the weak scaling data we can estimate a parallel efficiency above 50% for a system with 100 million atoms.
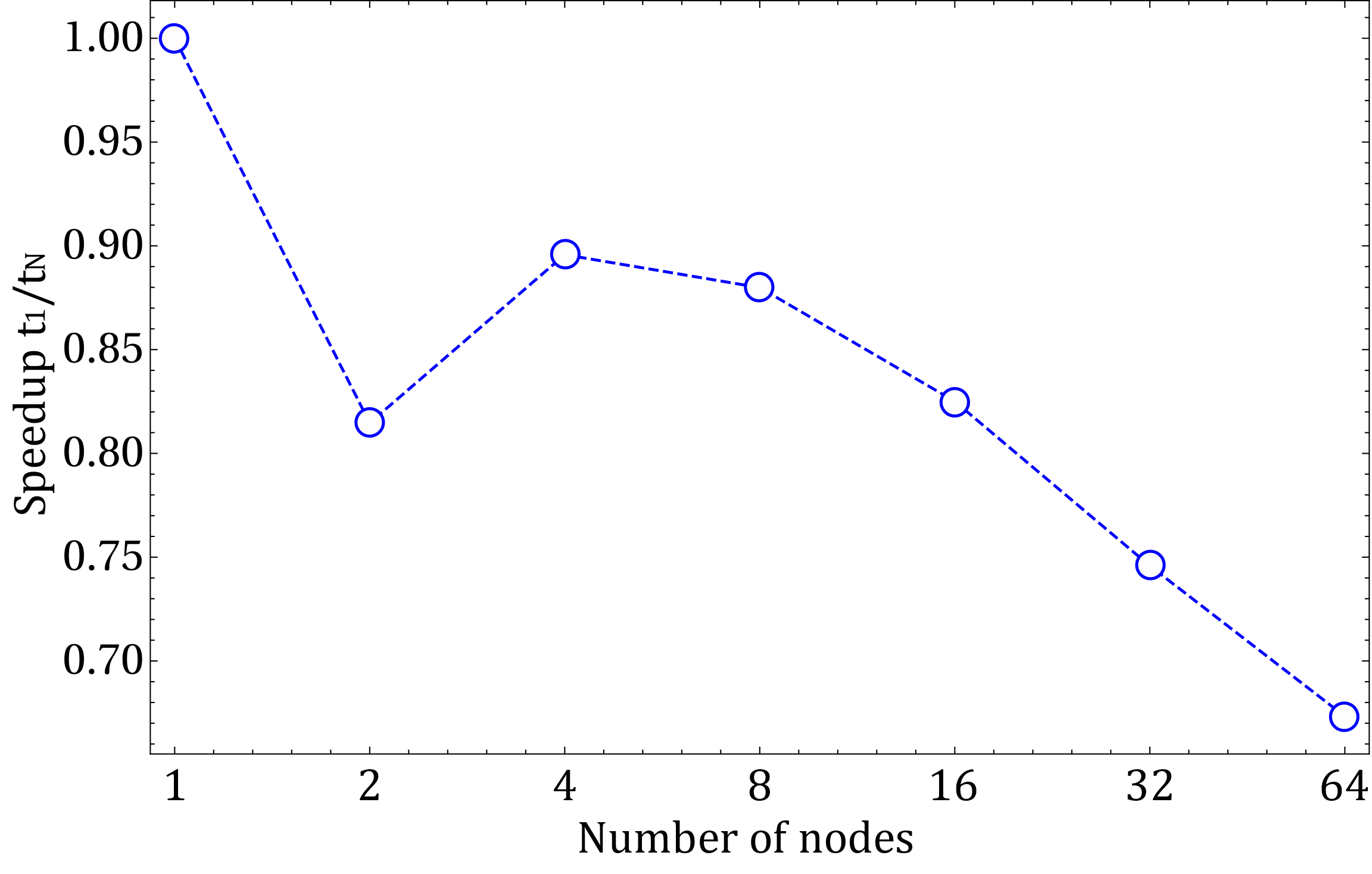
Figure 2. Parallel efficiency when the size of the systems scales linear with number of nodes (0.25 million atoms on each node)
Summary:
The strong scaling shows that on N GPU nodes systems with N-2N million atoms run efficiently. The weak scaling shows that efficiency will drop below 50% over 100 N. So, from these tests an optimal system up to 100 N should be of the order of 100's million of atoms.
References:
[1] Russo, M. F. Jr & van Duin, A. C. T. Atomistic-scale simulations of chemical reactions: Bridging from quantum chemistry to engineering. Nucl. Instrum. Methods Phys. Res. B 269, 1549–1554 (2011).
Download pdf version
Lammps reax benchmark on jewels