 Source code
Source code
MAELAS code is a software to calculate anisotropic magnetostriction coefficients and magnetoelastic constants up to second order. It generates required input files for VASP code to perform Density Functional Theory calculations, and it deduces the value of magnetostriction coefficients from the calculated energies given by VASP. If the elastic tensor is provided, then it can also calculate the magnetoelastic constants.
MAELAS can also be used with other DFT codes instead of VASP, after file conversion to VASP format files.
 Source code
Source code
JorGπ [Georgie Pie] is an automatic scheme of mapping an ab-initio system (so far using VASP) onto Heisenberg model
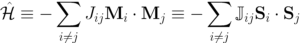 .
.
It provides utilities for generation and post processing of magnetic structures.
Code description
MAELAS
usage: maelas [-h] [-i POS] [-n NDIST] [-s STRAIN] [-k KP] [-g] [-d] [-r] [-m]
[-s1 SPIN1 SPIN1 SPIN1] [-s2 SPIN2 SPIN2 SPIN2] [-b] [-e ELAS]
[-sp SYMPRE] [-sa SYMANG] [-sg SG0] [-c CORE] [-t TIME]
[-f VASP_FOLD] [-mp MPI] [-a P_ID] [-l LOAD_MODULE] [-q QUEUE]
MAELAS code v1.0
optional arguments:
-h, --help show this help message and exit
-i POS Name of the initial non-distorted POSCAR file
(default: POSCAR)
-n NDIST Number of distorted states for each magnetostriction
mode (default: 7)
-s STRAIN Maximum value of the parameter epsilon for the strain
tensor to generate the distorted POSCAR files
(default: 0.01)
-k KP VASP automatic k-point mesh generation to create the
KPOINTS file (default: 60)
-g Generation of required VASP files for the calculation
of magnetostriction coefficients. Notation of the
generated output files: POSCAR_A_B (volume-conserving
distorted cell where A=magnetostriction mode,
B=distorted cell), INCAR_A_C (non-collinear
calculation where A=magnetostriction mode, C=spin
orientation case), INCAR_std (collinear calculation).
How to run the VASP calculations: For each generated
POSCAR_A_B one should run first a collinear
calculation using INCAR_std and use the generated
WAVECAR and CHGCAR files to run non-collinear
calculations for each INCAR_A_C using the same
POSCAR_A_B. It also generates bash scripts to run VASP
calculations easily (vasp_maelas, vasp_jsub, vasp_0)
and to get calculated OSZICAR_A_B_C files
(vasp_cp_oszicar)
-d Derivation of magnetostriction coefficients from the
energy written in the OSZICAR files. WARNING!: OSZICAR
files must be in the same folder where you run MAELAS
using the notation OSZICAR_A_B_C obtained for
POSCAR_A_B and INCAR_A_C. Distorted POSCAR files
(POSCAR_A_B) must be in this folder too (jointly with
the initial non-distorted POSCAR which should be
specified using tag -i). Specify the number of
distorted states to be considered in the calculation
of magnetostriction coefficients using tag -n. Energy
values extracted from OSZICAR_A_B_C files are shown in
files ene_A_C.dat and fit_ene_A_C.png. The energy
difference between the two spin configurations for
each magnetostriction mode are shown in Figs. dE_A.png
-r Generation of required VASP files for the cell
relaxation
-m Generation of required VASP files to test MAE
-s1 SPIN1 SPIN1 SPIN1
First spin direction to calculate MAE: s1x s1y s1z
-s2 SPIN2 SPIN2 SPIN2
Second spin direction to calculate MAE: s2x s2y s2z
-b Calculation of the magnetoelastic constants from the
calculated magnetostriction coefficients and provided
elastic tensor. For this option the tag -d must be
included as well as tag -e with the elastic tensor
file
-e ELAS File with the elastic tensor data in the same format
and units (GPa) as it is written by ELAS code (file
ELADAT). You can check this format in the Examples
folder
-sp SYMPRE Tolerance for symmetry finding (default: 0.01)
-sa SYMANG Angle tolerance for symmetry finding (default: 5.0)
-sg SG0 Space group number 1-230. If it is equal to 0, then it
will be determined by a symmetry analysis (default: 0)
-c CORE Number of cores for the VASP calculation (default: 24)
-t TIME Number of maximum CPU hours for the VASP calculation
(default: 48)
-f VASP_FOLD Folder where you will run VASP calculations (default:
/scratch)
-mp MPI Command for mpi run of VASP (default: mpiexec.hydra)
-a P_ID Project id for running jobs in HPC facilities
(default: OPEN-X-X)
-l LOAD_MODULE Module of VASP that should be loaded (default:
VASP/5.4.4-intel-2017c-mkl=cluster)
-q QUEUE Type of queue to be used for VASP calculations in HPC
facilities (default: qprod)JorGπ
JorGpi-startup
usage: JorGpi-startup [-h] [--input INPUT] [--incar INCAR] [--output OUTPUT]
[--cutOff CUTOFF | --neighbor NEIGHBOR]
[--Wyckoffs WYCKOFFS] [--reference REFERENCE]
[--elements ELEMENTS]
[--group {1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18} [{1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18} ...]]
[--period {2p,3p,4p,5p,6p,3d,4d,5d,4f,5f} [{2p,3p,4p,5p,6p,3d,4d,5d,4f,5f} ...]]
[--block {P,D,F} [{P,D,F} ...]] [--symmetry] [--redundant]
[--spin-orbit] [--refined]
[--extra-dimentions EXTRA-DIMENTIONS]
Find minimal number of unique spin-flips
optional arguments:
-h, --help show this help message and exit
--input INPUT, -i INPUT
input POSCAR file
--incar INCAR, --INCAR INCAR, -I INCAR
input INCAR file
--output OUTPUT, -o OUTPUT
output directory
--cutOff CUTOFF, -R CUTOFF
a cut-off distance (in Å) for calculations
--neighbor NEIGHBOR, -N NEIGHBOR
a rank of the last Neighbor taken into account
--Wyckoffs WYCKOFFS, -W WYCKOFFS
narrows down the atomic selection to the atoms in
positions defined by string (eg. 'abc')
--reference REFERENCE, -r REFERENCE
number of reference atom in inputFile
--elements ELEMENTS, -E ELEMENTS
string of all elements taken into account (eg. 'CuO')
--group {1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18} [{1,2,3,4,5,6,7,8,9,10,11,12,13,14,15,16,17,18} ...]
group number (eg. 1 <=> 'HLiNaKRbCsFr')
--period {2p,3p,4p,5p,6p,3d,4d,5d,4f,5f} [{2p,3p,4p,5p,6p,3d,4d,5d,4f,5f} ...]
period name (eg. 3d <=>
'$Sc$Ti$V$Cr$Mn$Fe$Co$Ni$Cu$Zn$')
--block {P,D,F} [{P,D,F} ...]
block name (eg. P <=> '$B$C$N$O$F$Al$Si$P$S$Cl$Ga$Ge$A
s$Se$Br$In$Sn$Sb$Te$I$Tl$Pb$Bi$Po$At$')
--symmetry, -S symmetry run only (default False)
--redundant creates a redundant system of equations for final
calculation of the Heisenberg exchange interaction
(default False)
--spin-orbit, --SOC (work-in-progress) is sping-orbit coupling enabled
(default False)
--refined should use refined supercell (default False)
--extra-dimentions EXTRA-DIMENTIONS, -X EXTRA-DIMENTIONS
string "X Y Z" of extra cell copies in each directions
(eg. "0 0 1")JorGpi-pickup
usage: JorGpi-pickup [-h] [--number-of-interactions #J] --reference dir
[--units {eV,meV,Ry,mRy,He,mHe,K}] --elements symbol
[symbol ...] --directories dir [dir ...]
Finding Js
optional arguments:
-h, --help show this help message and exit
--number-of-interactions #J, -J #J
number of exchange-interaction magnitudes to-be-
included in calculations
--reference dir, --noFlip dir, -R dir
reference directory (usually noFlip/)
--units {eV,meV,Ry,mRy,He,mHe,K}, -U {eV,meV,Ry,mRy,He,mHe,K}
units of energy
--elements symbol [symbol ...], --atoms symbol [symbol ...], -E symbol [symbol ...]
Symbol of elements taken into account in calculations
--directories dir [dir ...], -D dir [dir ...]
Directories containing flipped configurations (eg.
flip00000/)